New study exploiting viral DNA genomes to explore the dispersal history of African swine fever lineages in Europe
Published on May 29, 2025, by Fabiana Gámbaro & Simon Dellicour
African swine fever virus (ASFV) is a highly virulent DNA virus that causes African swine fever, a severe hemorrhagic disease affecting domestic and wild pigs, leading to significant animal health burdens and economic losses. Initially limited to the sub-Saharan African region, ASFV genotype II has spread globally and is now a major concern in Africa, Europe, Asia, the Pacific and, more recently, the Caribbean. In this study, we performed phylogenetic and phylogeographic analyses using newly sequenced ASFV genomes from Lithuania, combined with previously available complete genomes, to investigate the spatiotemporal dispersal dynamics of ASFV genotype II in Europe. Our analysis suggests that ASFV genotype II has not been recently imported to Europe from other regions; instead, the spread is largely driven by long-distance dispersal, followed by regional (within-country) circulation. The estimated dispersal metrics suggest that ASFV has a slower dispersion capacity compared to other pig-transmitted viruses and is associated with a notable degree of spatial structure. Despite these findings, significant uncertainty remains regarding certain ancestral locations, highlighting challenges related to applying phylodynamic methods to DNA viruses with low genetic variability. Nevertheless, in our study we managed to implement a phylogeographic framework to investigate major patterns of ASFV dispersion in Europe and the contribution of international importations in the establishment of regional transmission chains. This framework could be further expanded as more genomes become available. Our study emphasises the need for increased genomic surveillance to enlarge the ASFV genome database to support outbreak control. Read the whole study here.
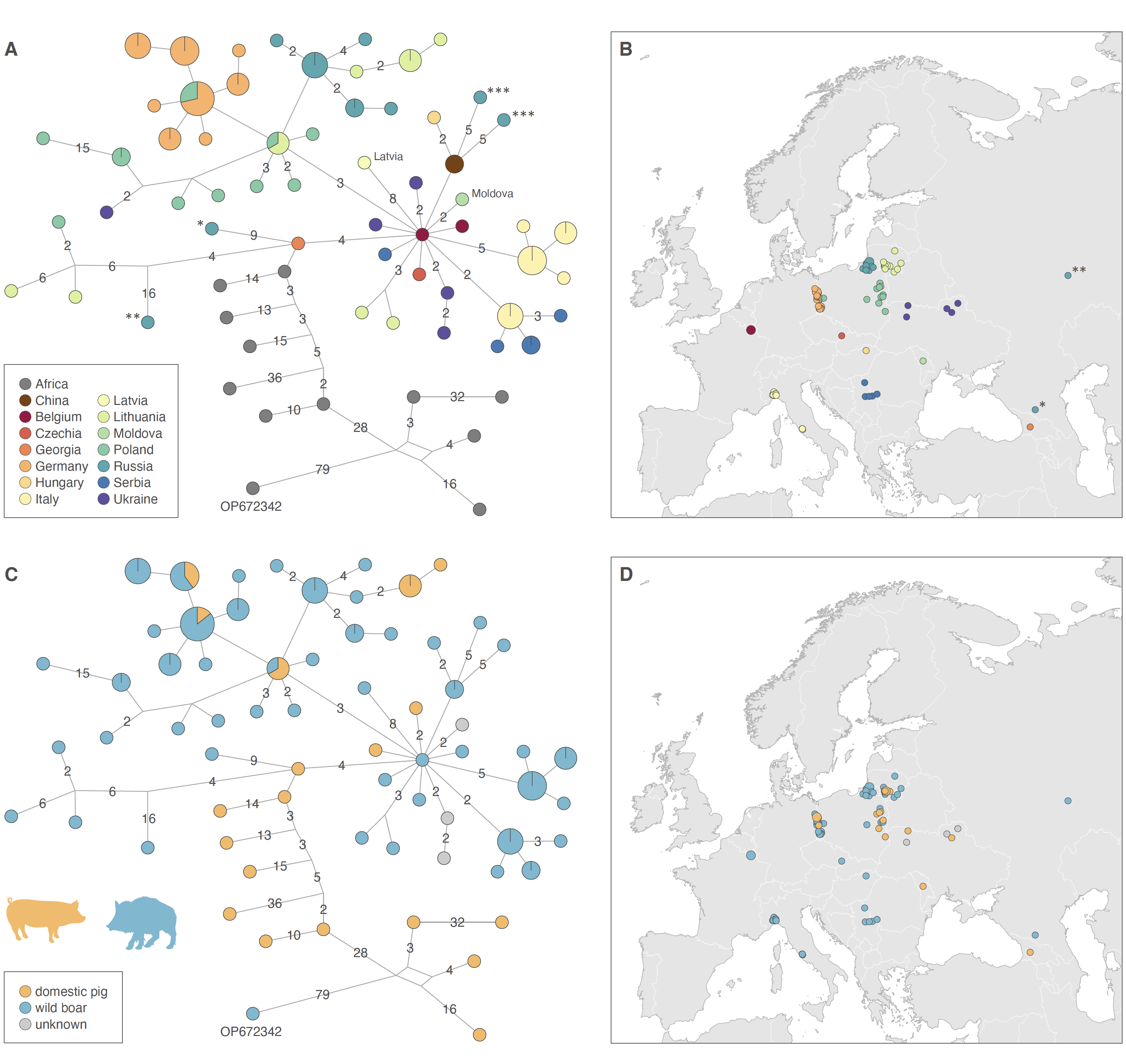 Figure: genetic variability (A,C) and sampling maps (B,D) of ASFV whole genomes. In both haplotype networks (A, C), each haplotype corresponds to a unique sequence represented by a circle, the size of which is proportional to its overall sampling frequency, and the genetic relatedness between haplotypes are represented by line segments, the numbers reported on line segments indicating the number of mutational changes separating two haplotypes. The two haplotype networks are identical but coloured according to distinct sampling information: while the first haplotype network is coloured according to the country of origin of each genomic sequence (A), the second haplotype network is coloured according the host type of origin (domestic pig or wild boar) of the different samples (B). Within both networks, we indicated the position of sample OP672342 from Nigeria as it was identified as a recombinant sample discarded from the alignment for subsequent analyses. In the first (B) and second (D) sampling maps, the origin of each genomic sample is coloured according to the country of origin and the host type, respectively. (*) and (**) refer to two genomic sequences coming from Kabardino-Balkaria and Ulyanovsk in Russia, respectively; and (***) to two genomic sequences collected in Armur and Primorsky in the Russian Far-East.
Figure: genetic variability (A,C) and sampling maps (B,D) of ASFV whole genomes. In both haplotype networks (A, C), each haplotype corresponds to a unique sequence represented by a circle, the size of which is proportional to its overall sampling frequency, and the genetic relatedness between haplotypes are represented by line segments, the numbers reported on line segments indicating the number of mutational changes separating two haplotypes. The two haplotype networks are identical but coloured according to distinct sampling information: while the first haplotype network is coloured according to the country of origin of each genomic sequence (A), the second haplotype network is coloured according the host type of origin (domestic pig or wild boar) of the different samples (B). Within both networks, we indicated the position of sample OP672342 from Nigeria as it was identified as a recombinant sample discarded from the alignment for subsequent analyses. In the first (B) and second (D) sampling maps, the origin of each genomic sample is coloured according to the country of origin and the host type, respectively. (*) and (**) refer to two genomic sequences coming from Kabardino-Balkaria and Ulyanovsk in Russia, respectively; and (***) to two genomic sequences collected in Armur and Primorsky in the Russian Far-East.
Reference: Gámbaro F, Goatley LC, Foster TJ, Tennakoon C, Freimanis GL, Van Borm S, Masiulis M, Bušauskas P, Netherton CL, Dellicour S (2025). Exploiting viral DNA genomes to explore the dispersal history of African swine fever genotype II lineages in Europe. Genome Biology & Evolution 17: evaf102