New study in PNAS on the comparative performance of novel viral landscape phylogeography approaches
Published on June 25, 2025, by Simon Dellicour
The rapid evolution of RNA viruses implies that their evolutionary and ecological processes occur on the same time scale. Genome sequences of these pathogens therefore can contain information about the processes that govern their transmission and dispersal. Landscape phylogeographic approaches use phylogeographic reconstructions to investigate the impact of environmental factors and variables on the spatial spread of viruses. In this new study, we extend and improve existing approaches and develop three novel landscape phylogeographic methods that can test the impact of continuous environmental factors on the diffusion velocity of viral lineages. In order to evaluate the different methods, we also implemented two simulation frameworks to test and compare their statistical performance. The results enable us to formulate clear guidelines for the use of three complementary landscape phylogeographic approaches that have sufficient statistical power and low rates of false positives. Our open-source methods are available to the scientific community and can be used to investigate the drivers of viral spread, with potential benefits for understanding virus epidemiology and designing tailored intervention strategies. Read the whole study here.
R scripts related to the analyses based on simulated datasets are all available, along with the associated input and output files, on GitHub. Continuous phylogeographic simulations and landscape phylogeographic analyses were conducted using new functions implemented in the R package “seraphim”, which includes new tutorials dedicated to (i) the phylogeographic simulations conducted on environmental rasters and (ii) the prior-informed landscape phylogeographic analyses based on an MDS or cartogram transformation, as well as (iii) an updated tutorial dedicated to the post hoc landscape phylogeographic analyses on the impact of environmental factors on the diffusion velocity of viral lineages (the latest tutorial also including the isolation-by-resistance analyses).
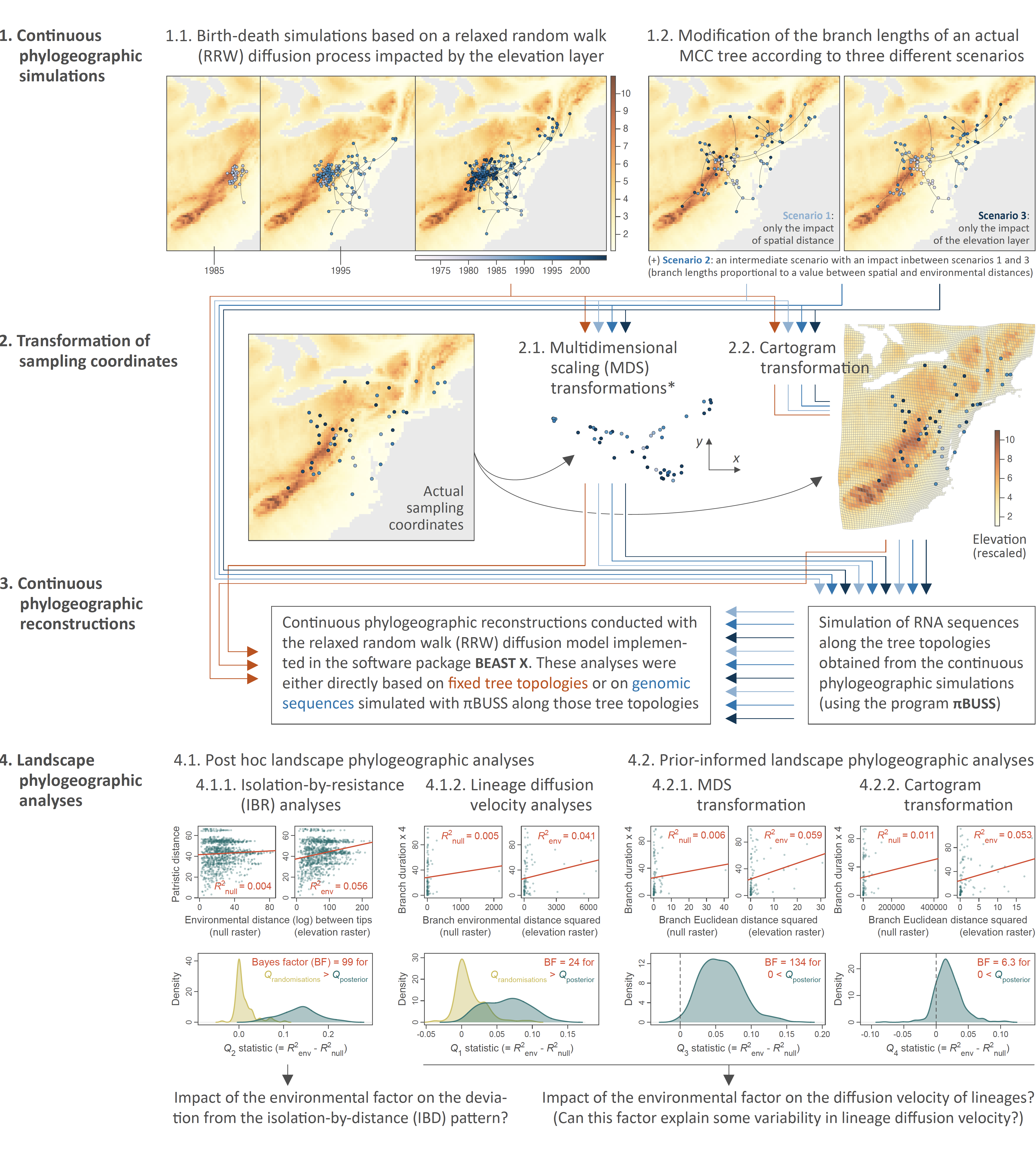 Simulation and analytical workflow implemented to assess the statistical performance of four landscape phylogeographic approaches to test the impact of environmental factors on the diffusion velocity of viral lineages or on the deviation from an isolation-by-distance pattern. We here assessed the performance of two post hoc (isolation-by-resistance and lineage diffusion velocity analyses) and two prior-informed landscape phylogeographic approaches (analyses of continuous phylogeographic reconstructions based on sampling coordinates obtained either after a multidimensional scaling or cartogram transformation). Continuous phylogeographic simulations (step 1), transformations of sampling coordinates (step 2), and landscape phylogeographic analyses were conducted with the R package “seraphim”, the RNA sequences simulations with the program πBUSS, and the continuous phylogeographic reconstructions (step 3) with the software package BEAST X version 1.10.5 (19). (*) Multidimensional scaling (MDS) transformations were conducted based on environmental distances among pair of tip nodes, which were computed using either the least-cost (5) or Circuitscape (3, 32) path model while either considering the environmental raster (here an elevation raster with values rescaled between 0 and 10) or a corresponding “null” raster with accessible raster cell values uniformly equal to “1” (see the text for further detail). “MCC tree” refers to a maximum clade credibility tree.
Simulation and analytical workflow implemented to assess the statistical performance of four landscape phylogeographic approaches to test the impact of environmental factors on the diffusion velocity of viral lineages or on the deviation from an isolation-by-distance pattern. We here assessed the performance of two post hoc (isolation-by-resistance and lineage diffusion velocity analyses) and two prior-informed landscape phylogeographic approaches (analyses of continuous phylogeographic reconstructions based on sampling coordinates obtained either after a multidimensional scaling or cartogram transformation). Continuous phylogeographic simulations (step 1), transformations of sampling coordinates (step 2), and landscape phylogeographic analyses were conducted with the R package “seraphim”, the RNA sequences simulations with the program πBUSS, and the continuous phylogeographic reconstructions (step 3) with the software package BEAST X version 1.10.5 (19). (*) Multidimensional scaling (MDS) transformations were conducted based on environmental distances among pair of tip nodes, which were computed using either the least-cost (5) or Circuitscape (3, 32) path model while either considering the environmental raster (here an elevation raster with values rescaled between 0 and 10) or a corresponding “null” raster with accessible raster cell values uniformly equal to “1” (see the text for further detail). “MCC tree” refers to a maximum clade credibility tree.
Reference: Dellicour S, Gámbaro F, Jacquot M, Lequime S, Baele G, Gilbert M, Pybus OG, Suchard MA, Lemey P (2025). Comparative performance of viral landscape phylogeography approaches. Proceedings of the National Academy of Sciences of the USA 122: e2506743122