New review: accommodating sampling location uncertainty in continuous phylogeography
Published on July 07, 2022, by Simon Dellicour
Phylogeographic inference of the dispersal history of viral lineages offers key opportunities to tackle epidemiological questions about the spread of fast-evolving pathogens across human, animal, and plant populations. In continuous space, i.e. when locations are specified by longitude and latitude, these reconstructions are however often limited by the availability or accessibility of precise sampling locations required for such spatially-explicit analyses. In our new study published in Virus Evolution, we review the different approaches that can be considered when genomic sequences are associated with a geographic area of sampling instead of precise coordinates. In particular, we describe and compare the approaches to define homogeneous and heterogeneous prior ranges of sampling coordinates. Read the whole study here.
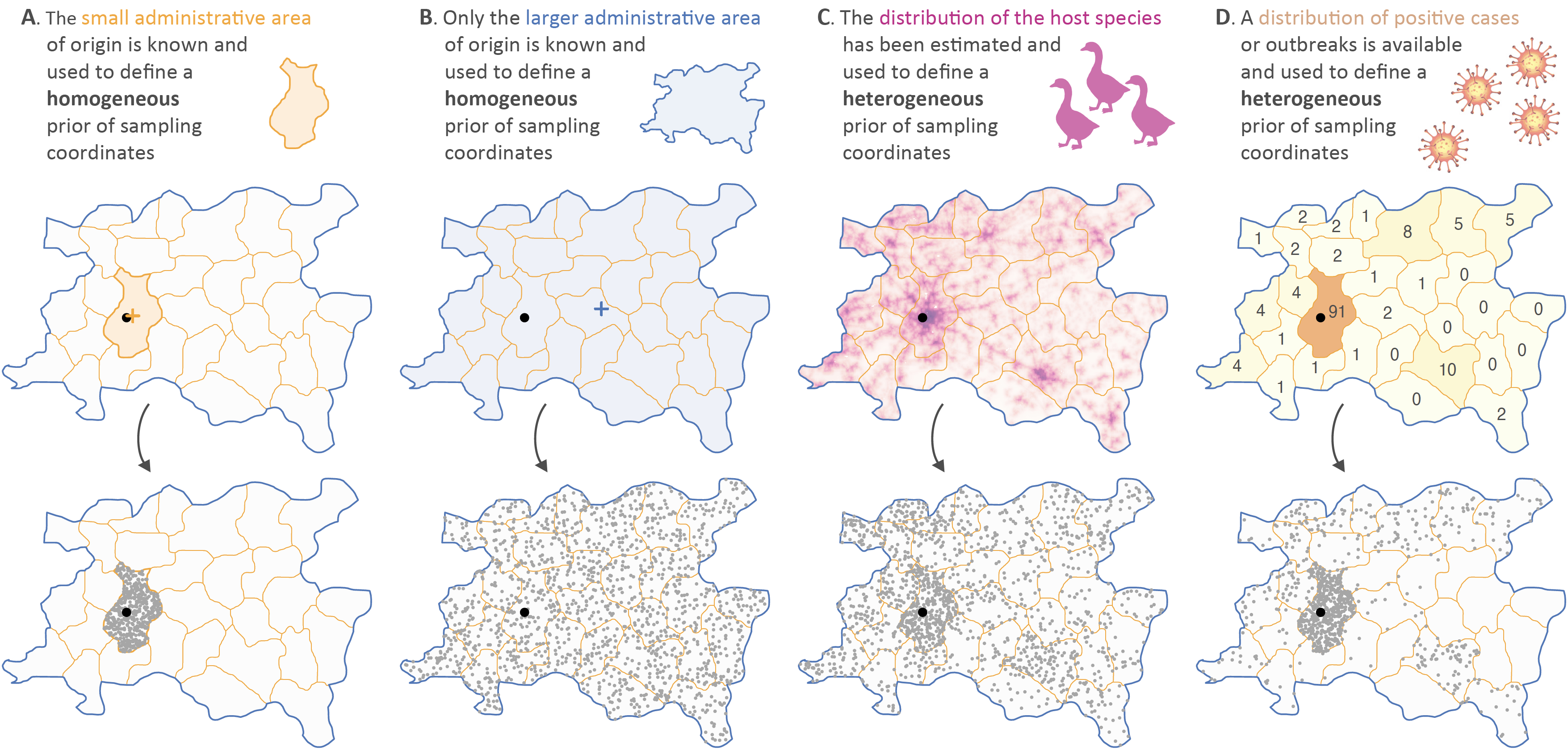 Figure: illustration of the different procedures that can be used to define a homogeneous or heterogeneous prior range of sampling coordinates. To perform a continuous (i.e. spatially-explicit) phylogeographic reconstruction of the dispersal history of a fast-evolving pathogen, geographic coordinates associated with the sampling location of each genomic sequence included in the analysis are required. However, precise sampling locations are frequently unknown, not available or not accessible in the case of human cases protected by privacy data protection rules. When the small administrative area of origin is known (A), sampling coordinates can be integrated through a homogeneous prior range delineated by the polygon of this administrative area. On the contrary, when only the upper-level larger administrative area (e.g. province, state, etc.) is known (B), it becomes less relevant to consider the associated polygon to define the prior range of sampling coordinates. In the latter case, and in order to avoid having to discard the considered sample from the data set, external data can be used to define an heterogeneous prior range of sampling coordinates, which thus uses prior information to decrease the uncertainty associated with the geographic origin of the sample. Practically, two different types of external data can be considered: host species distribution (C) or, ideally, the spatial repartition of positive or outbreak cases recorded at the considered sampling time (D). In both cases, those external data are used to define the relative sampling probability assigned to a series of smaller polygon units. In all maps, the actual but unknown sampling point is indicated by a black dot. In panels A and B, the centroid point of the small and larger administrative area of origin is displayed as an orange and blue cross, respectively.
Figure: illustration of the different procedures that can be used to define a homogeneous or heterogeneous prior range of sampling coordinates. To perform a continuous (i.e. spatially-explicit) phylogeographic reconstruction of the dispersal history of a fast-evolving pathogen, geographic coordinates associated with the sampling location of each genomic sequence included in the analysis are required. However, precise sampling locations are frequently unknown, not available or not accessible in the case of human cases protected by privacy data protection rules. When the small administrative area of origin is known (A), sampling coordinates can be integrated through a homogeneous prior range delineated by the polygon of this administrative area. On the contrary, when only the upper-level larger administrative area (e.g. province, state, etc.) is known (B), it becomes less relevant to consider the associated polygon to define the prior range of sampling coordinates. In the latter case, and in order to avoid having to discard the considered sample from the data set, external data can be used to define an heterogeneous prior range of sampling coordinates, which thus uses prior information to decrease the uncertainty associated with the geographic origin of the sample. Practically, two different types of external data can be considered: host species distribution (C) or, ideally, the spatial repartition of positive or outbreak cases recorded at the considered sampling time (D). In both cases, those external data are used to define the relative sampling probability assigned to a series of smaller polygon units. In all maps, the actual but unknown sampling point is indicated by a black dot. In panels A and B, the centroid point of the small and larger administrative area of origin is displayed as an orange and blue cross, respectively.
Reference: Dellicour S, Lemey P, Suchard MA, Gilbert M, Baele G (2022). Accommodating sampling location uncertainty in viral phylogeography. Virus Evolution 8: veac041