New study in Cell on the recency and spatial origins of the bat viruses ancestral to SARS-CoV and SARS-CoV-2
Published on May 07, 2025, by Simon Dellicour
The emergence of SARS-CoV in 2002 and SARS-CoV-2 in 2019 led to increased sampling of sarbecoviruses circulating in horseshoe bats. In a new comprehensive study, we employed phylogenetic inference while accounting for recombination of bat sarbecoviruses and found that the closest-inferred bat virus ancestors of SARS-CoV and SARS-CoV-2 existed less than a decade prior to their emergence in humans. Phylogeographic analyses showed bat sarbecoviruses traveled at rates approximating their horseshoe bat hosts and circulated in Asia for millennia. We found that the direct ancestors of SARS-CoV and SARS-CoV-2 are unlikely to have reached their respective sites of emergence via natural dispersal in the bat reservoir alone, though interactions with intermediate hosts through wildlife trade likely played a role in zoonotic spillover. These results can guide future sampling efforts and demonstrate that viral genomic fragments extremely closely related to SARS-CoV and SARS-CoV-2 were circulating in horseshoe bats, confirming their importance as the reservoir species for SARS viruses. Read the whole study here.
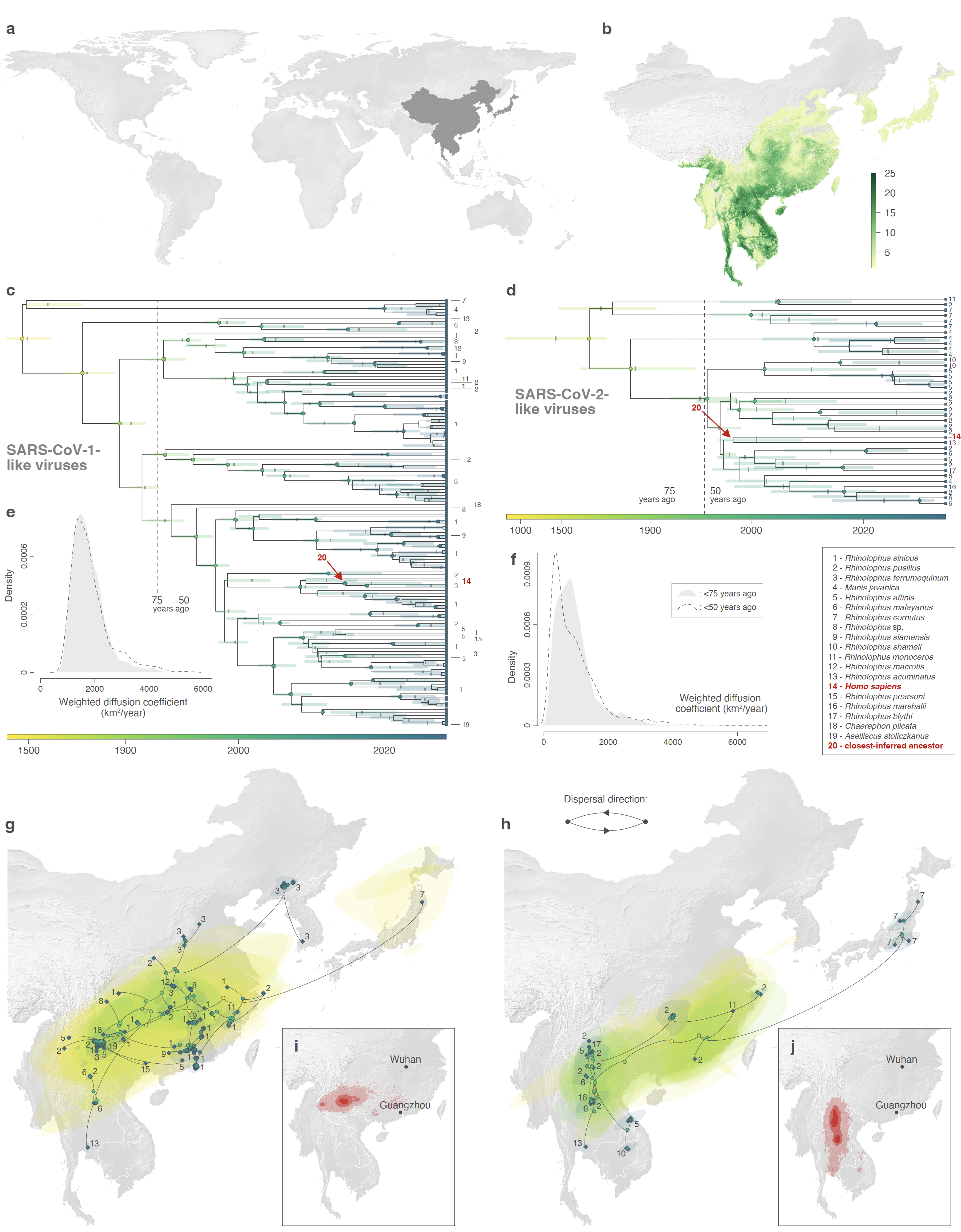 Figure: estimated evolutionary history of SARS-CoV-1-like (left) and SARS-CoV-2-like (right) viruses. a: global map with the study area highlighted in dark gray. b Rhinolophid (horseshoe bat) species richness with the scale bar showing the number of co-occurring Rhinolophid species per km2. c-d: time-scaled maximum clade credibility (MCC) phylogenies of individual non-recombinant regions (NRRs; NRR14 for SARS-CoV-1-like viruses, and NRR37 for SARS-CoV-2-like viruses) obtained after correction for substitution saturation, shown on a log-transformed time scale. Horizontal segments displayed at each internal node indicate the 95% HPD interval of the estimated node age, with a light grey vertical bar indicating the median node age. The closest-inferred ancestors of SARS-CoV-1 and SARS-CoV-2 are indicated by red arrows. e-f: weighted diffusion coefficient estimates based on phylogenetic branches occurring less than 75 (plain light gray curves) and 50 (dashed curves) years ago. g-h: reconstruction of the dispersal history of viral lineages for two NRRs (NRR14 for SARS-CoV-1-like viruses, and NRR37 for SARS-CoV-2-like viruses) obtained through continuous phylogeographic inference (see Figures S10-S14 for all other NRRs). Nodes are coloured from yellow (oldest most recent common ancestor) to blue (most recent collection date) and are superimposed on 80% HPD polygons coloured according to the same log-transformed time scale, reflecting the uncertainty of the Bayesian phylogeographic inference. 80% HPD polygons were only computed and reported for the last 1,000 years. Dispersal direction (anti-clockwise) of viral lineages is indicated by the edge curvature. In panels c-d-g-h, tip nodes are displayed as squares and are associated with a number indicating the host species from which the virus was sampled; internal nodes are displayed as circles. The host species are numbered by decreasing order of sampling frequency. i-j estimated position of the closest-inferred bat virus ancestors of SARS-CoV-1 (i) and SARS-CoV-2 (j) based on the continuous phylogeographic reconstruction of all NRRs. The 95%, 75% and 50% HPD regions are displayed with an increasing red color darkness. Note that we use the PoW-transformed trees to illustrate the overall time-scale of the evolutionary histories, but refer to Figure 1 and Supplementary Tables 4 and 5 for estimates of the time of the closest-inferred ancestor.
Figure: estimated evolutionary history of SARS-CoV-1-like (left) and SARS-CoV-2-like (right) viruses. a: global map with the study area highlighted in dark gray. b Rhinolophid (horseshoe bat) species richness with the scale bar showing the number of co-occurring Rhinolophid species per km2. c-d: time-scaled maximum clade credibility (MCC) phylogenies of individual non-recombinant regions (NRRs; NRR14 for SARS-CoV-1-like viruses, and NRR37 for SARS-CoV-2-like viruses) obtained after correction for substitution saturation, shown on a log-transformed time scale. Horizontal segments displayed at each internal node indicate the 95% HPD interval of the estimated node age, with a light grey vertical bar indicating the median node age. The closest-inferred ancestors of SARS-CoV-1 and SARS-CoV-2 are indicated by red arrows. e-f: weighted diffusion coefficient estimates based on phylogenetic branches occurring less than 75 (plain light gray curves) and 50 (dashed curves) years ago. g-h: reconstruction of the dispersal history of viral lineages for two NRRs (NRR14 for SARS-CoV-1-like viruses, and NRR37 for SARS-CoV-2-like viruses) obtained through continuous phylogeographic inference (see Figures S10-S14 for all other NRRs). Nodes are coloured from yellow (oldest most recent common ancestor) to blue (most recent collection date) and are superimposed on 80% HPD polygons coloured according to the same log-transformed time scale, reflecting the uncertainty of the Bayesian phylogeographic inference. 80% HPD polygons were only computed and reported for the last 1,000 years. Dispersal direction (anti-clockwise) of viral lineages is indicated by the edge curvature. In panels c-d-g-h, tip nodes are displayed as squares and are associated with a number indicating the host species from which the virus was sampled; internal nodes are displayed as circles. The host species are numbered by decreasing order of sampling frequency. i-j estimated position of the closest-inferred bat virus ancestors of SARS-CoV-1 (i) and SARS-CoV-2 (j) based on the continuous phylogeographic reconstruction of all NRRs. The 95%, 75% and 50% HPD regions are displayed with an increasing red color darkness. Note that we use the PoW-transformed trees to illustrate the overall time-scale of the evolutionary histories, but refer to Figure 1 and Supplementary Tables 4 and 5 for estimates of the time of the closest-inferred ancestor.
Reference: Pekar JE, Lytras S, Ghafari M, Magee AF, Parker E, Wang Y, Ji X, Havens JL, Katzourakis A, Vasylyeva TI, Suchard MA, Hughes AC, Hughes J, Rambaut A, Robertson DL*, Dellicour S*, Worobey M*, Wertheim JO*, Lemey P* (2025). The recency and geographical origins of the bat viruses ancestral to SARS-CoV and SARS-CoV-2. Cell, in press. (*) denotes equal contribution